
Estrogen receptor mutations and their role in breast cancer progression
Prasanna G Alluri, Corey Speers and Arul M Chinnaiyan
Breast Cancer Research 2014, 16:494 doi:10.1186/s13058-014-0494-7
Background
About 75% of breast cancers are hormone positive, and patients with these cancers, which are ER+ and usually also PR+, benefit from hormone therapy, which reduces estrogens by a variety of different means. Hormone therapy, for patients with hormone positive tumors, is often the most effective part of their therapy.
The bad news is that now that we have quite a few years of experience with hormone therapy, it can be seen that it isn’t a cure for everyone. One study showed that one-third of women treated with tamoxifen for 5 years have a breast cancer recurrence within 15 years. The question is why? Why does it work for some and not for others? What happens in the cancer cells of those who have recurrence and can we come up with a treatment if we understand the mechanism? This review explores this issue.
Findings
This paper is a review, meaning that it focuses on summarizing the state of the field rather than on presenting primary research data, although these authors do include some. Because it is a review, many of the statements made in the paper are based on other studies, and are referenced as such. So the findings are really a summary of the work of many labs.
The key finding is that ESR1 mutations can be found in metastatic recurrences of breast cancer, post-hormone therapy, with high frequency (perhaps as many as half of such patients). Such mutations are not found in treatment-naive, primary cancers, This suggests strongly that the mutations are selected for in cancers undergoing hormone therapy, and represent a mechanism for escaping this therapy.
Interestingly the vast majority of mutations in ESR1 are found in the ligand binding domain of the receptor. This is the part of the protein where the estrogen binds to activate the receptor. You can imagine a key (estrogen) entering a lock (the ligand binding domain) and turning it to reveal the activation domain (the bolt).
Even more interesting, most mutations cluster in a specific part of the ligand binding domain, a short distance from where the estrogen actually binds – residues 534-538. I have marked this region with yellow boxes in the image, which is a screenshot of the Weizmann Institute’s excellent first look tool for structure – you can see and experiment with the structure of the ligand binding domain here. In this cartoon image of the ligand binding domain more ordered structures are shown as curled arrow ribbons (alpha helices) while more flexible parts are shown as thinner, string like sections. The mutations appear to cluster in a small alpha helix, separated by the main part of the ligand binding domain by a flexible section.

ESR1 ligand binding domain (PDB file 1A52) on First Glance by the Weizmann Institute – http://oca.weizmann.ac.il/oca-docs/fgij/help.htm?licenseHelp
Interestingly, the region includes a phosphorylation site (537) which may indicate that changes in charge and structure here are regulatory. In fact, structural and functional analyses of mutations in this region suggest that such mutants mimic the structure of the activated receptor, and promote interaction with SRC-1, a powerful kinase that collaborates with ESR1 (and many other signaling molecules).
So it would seem that these mutations affect the way the ESR1 functions. It appears that these mutations make it possible for ESR1 to be active even when ligand is absent – this would allow the breast cancer cells to resume growth even while the patient suppresses estrogen with hormone therapy. Indeed Alluri and colleagues report that:
“Interestingly, all ESR1 mutants showed robust constitutive activation of the ERE reporter, unlike the wild-type ESR1 that had little activity in the absence of estradiol.”
A reporter in this context is a tool that allows you to measure the activity of a gene regulator, such as ESR1. So, the mutants have acquired the ability to be active without estrogen.
Interestingly, and this is key, the mutant receptors remained susceptible to some kinds of hormone therapy.
Here we have to take a step back and consider that there are, broadly speaking, two ways to oppose estrogen’s activity in the body:
- With a molecule that competes with estrogen for its receptors, but does not activate it. Think of a key that goes in the keyhole, but cannot turn the lock, while preventing a functioning key from entering. This is anti-estrogen therapy, and is exemplified by tamoxifen
- By interrupting the production of estrogen, by inhibiting enzymes in the pathways that make it. Think of it as removing all the keys, by preventing them being made in the first place. This is estrogen deprivation therapy and is exemplified by aromatase inhibitors.
What Alluri and colleagues observed is that the mutant receptors remained susceptible to the anti-estrogen effects of tamoxifen. Perhaps tamoxifen can still bind the mutant, and keep it in an inactive state. Furthermore, it appears that the mutations mostly occur in patients who are receiving estrogen deprivation therapy.
Another interesting observation, which is not presented in detail, is that the genes activated by the mutants ESR1s are, in part, different from those that are activated by the wild-type receptor. ESR1 exerts its effects by activating dozens or even hundreds of downstream genes. Which ones will determine its effect, and knowing that mutation leads to different genes may reveal new pathways of interest or at least point to potential targets that can be aimed at therapeutically once an ESR1 mutation has occurred.
This figure, Fig. 1 from the paper, sums up the findings very well:
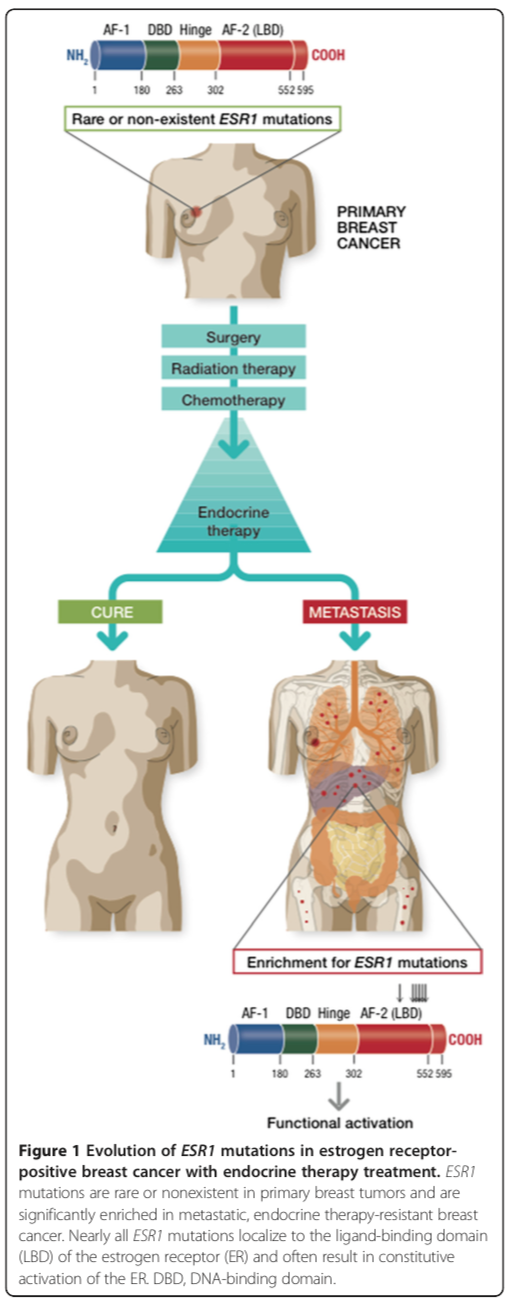
Alluri et al Figure 1
Comment
The most significant finding here is that an important element in some instances of the recurrence of hormone dependent breast cancer is mutation in ESR1, and that in such instances anti-estrogen therapy may be effective. However, it was already clinically known that women on aromatase inhibitors who experience a recurrence may well benefit from, say, tamoxifen. Now we have a biological explanation for some if not most of these cases.
Beyond that, the fact that SRC1 is involved might allow to future developments of combination therapies, or follow up therapies of a different nature. Similarly, the gene expression pattern differences between ESR1 mutants and wild type may well lead to new approaches, and biological understanding.
